


Chronic Granulomatous Disease (CGD) is a rare inherited immune disorder where phagocytes cannot kill certain bacteria and fungi, leading to recurrent infections and granuloma formation. Understanding its causes, symptoms, diagnosis, and management is essential in nursing and clinical practice.
Introduction
Chronic Granulomatous Disease (CGD) represents a group of rare, inherited disorders affecting the immune system’s ability to combat certain infections. First described in the 1950s, CGD has since become a subject of intense clinical and scientific investigation due to its profound implications for affected individuals and its role in advancing our understanding of innate immunity.
Definition and Epidemiology
What is Chronic Granulomatous Disease?
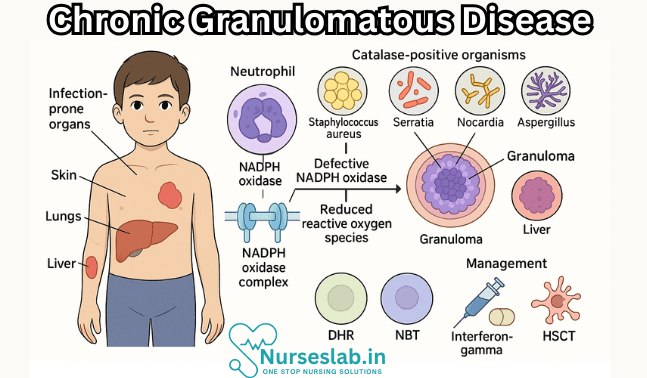
Chronic Granulomatous Disease is a primary immunodeficiency disorder characterised by the inability of phagocytes—especially neutrophils and macrophages—to effectively kill certain bacteria and fungi. This defect results from a malfunction in the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, leading to recurrent, life-threatening infections and granuloma formation in various tissues. The term “granulomatous” arises from the tendency of affected individuals to develop granulomas—organised collections of immune cells formed in response to persistent inflammation.
Prevalence and Demographics
CGD is considered a rare disease, with an estimated global incidence of approximately 1 in 200,000 to 250,000 live births. The prevalence may vary by region, with higher rates observed in populations with higher rates of consanguinity. CGD affects both males and females, although certain genetic forms are more prevalent in males. The disease is usually diagnosed in early childhood, often within the first few years of life, but milder cases may remain undetected until adolescence or adulthood.
Etiology and Genetics
Genetic Mutations and Inheritance Patterns
Chronic Granulomatous Disease is primarily caused by inherited mutations affecting the genes that encode components of the phagocyte NADPH oxidase complex. This multi-protein complex is essential for generating reactive oxygen species (ROS) during the “respiratory burst,” a process critical for intracellular killing of pathogens.
- X-linked CGD: The majority of CGD cases (about 65–70%) are due to mutations in the CYBB gene, which encodes the gp91phox protein. This form follows an X-linked recessive inheritance pattern, making it significantly more common in males. Female carriers may exhibit mild or no symptoms due to lyonisation (random X-chromosome inactivation).
- Autosomal Recessive CGD: The remaining cases result from autosomal recessive mutations in genes encoding other NADPH oxidase subunits, such as NCF1 (p47phox), NCF2 (p67phox), NCF4 (p40phox), and CYBA (p22phox). Both males and females are equally affected in these forms.
Types of CGD
- X-linked (gp91phox deficiency): Most common and generally associated with more severe disease.
- p47phox deficiency: Common autosomal recessive form, often seen in certain populations due to founder effects.
- p67phox and p22phox deficiencies: Less common autosomal recessive forms.
- p40phox deficiency: Rare and recently described, with variable clinical severity.
Pathophysiology
Mechanisms of Disease
The pathophysiology of CGD centres on defects in the NADPH oxidase complex of phagocytes. Under normal circumstances, this enzyme complex is activated upon ingestion of pathogens, leading to the generation of superoxide anions and other ROS. These molecules are toxic to many bacteria and fungi, playing a critical role in microbial killing. In CGD, mutations disrupt the assembly or function of NADPH oxidase, resulting in a defective respiratory burst and impaired ROS production.
Immune Dysfunction and Cellular Defects
The inability to generate ROS severely limits the capacity of neutrophils and macrophages to kill catalase-positive organisms. These pathogens are particularly problematic because they can degrade the hydrogen peroxide they produce, which might otherwise compensate for the host’s deficiency. Consequently, CGD patients are susceptible to persistent and recurrent infections. In response to chronic infection and inflammation, the immune system often forms granulomas—compact aggregates of macrophages and other immune cells—which can cause tissue damage and organ dysfunction.
Clinical Features
Common Symptoms
The clinical presentation of CGD is heterogeneous but typically includes recurrent bacterial and fungal infections, often beginning in infancy or early childhood. Key features include:
- Frequent, severe infections of the skin, lymph nodes, lungs, liver, and bones
- Delayed wound healing and persistent abscess formation
- Granuloma formation, leading to obstruction in the gastrointestinal or genitourinary tracts
- Failure to thrive in children, due to chronic illness
Types of Infections
CGD patients are particularly vulnerable to infections by catalase-positive bacteria and fungi, including:
- Staphylococcus aureus
- Serratia marcescens
- Burkholderia cepacia
- Nocardia species
- Aspergillus species (fungi)
These infections may present as pneumonia, lymphadenitis, hepatic abscesses, osteomyelitis, or sepsis. Unusual organisms or atypical clinical presentations should prompt consideration of CGD, especially in young patients with a history of recurrent or severe infections.
Complications
In addition to infections, CGD can lead to non-infectious complications such as:
- Obstructive granulomas in the gastrointestinal or urinary tract, leading to symptoms such as vomiting, abdominal pain, or urinary retention
- Chronic inflammation resulting in tissue fibrosis and organ dysfunction
- Autoimmune phenomena, including inflammatory bowel disease-like symptoms
Diagnosis
Laboratory Tests
The diagnosis of CGD rests on demonstrating impaired oxidative burst activity in phagocytes. Key laboratory tests include:
- Dihydrorhodamine (DHR) flow cytometry test: The gold standard for diagnosing CGD, this assay measures the ability of neutrophils to produce ROS in response to stimulation. A reduced or absent DHR signal is indicative of CGD.
- Nitroblue tetrazolium (NBT) test: An older method, now largely replaced by DHR, where a colour change indicates the production of superoxide. CGD neutrophils fail to reduce NBT, resulting in a negative test.
Imaging Studies
Radiological investigations may be warranted to evaluate the extent of infection or granuloma formation. Common modalities include:
- Chest X-ray or CT scan for pneumonia or lung abscesses
- Ultrasound or MRI for liver abscesses and lymphadenopathy
Genetic Testing
Molecular genetic analysis can identify the specific gene mutation responsible for CGD. This is essential for:
- Confirming the diagnosis, especially in atypical cases
- Determining the mode of inheritance (X-linked or autosomal recessive)
- Carrier detection and genetic counselling for affected families
Differential Diagnosis
CGD must be distinguished from other causes of recurrent infections and granulomatous inflammation, such as:
- Other primary immunodeficiencies (e.g., severe combined immunodeficiency, leukocyte adhesion deficiency)
- Acquired immunodeficiencies (e.g., HIV/AIDS)
- Chronic infections (e.g., tuberculosis)
- Autoimmune or inflammatory disorders
Management and Treatment
Antibiotic and Antifungal Prophylaxis
The foundation of CGD management is the prevention and prompt treatment of infections. This includes:
- Long-term antibiotic prophylaxis: Agents such as trimethoprim-sulphamethoxazole are commonly used to reduce the risk of bacterial infections.
- Antifungal prophylaxis: Itraconazole or similar agents are often prescribed to prevent fungal infections, particularly those caused by Aspergillus species.
Immunomodulation
Interferon-gamma (IFN-γ) therapy has been shown to enhance the microbicidal activity of phagocytes and reduce the frequency of infections in some patients. However, its use may be limited by side effects and variable efficacy.
Stem Cell Transplantation
Allogeneic haematopoietic stem cell transplantation (HSCT) offers the only curative treatment for CGD. Advances in transplantation techniques and supportive care have improved outcomes, especially when performed early in life and with a matched sibling donor. Risks include graft-versus-host disease, infections, and transplant-related morbidity.
Supportive Care
Comprehensive care for CGD patients involves:
- Prompt recognition and aggressive treatment of infections
- Surgical intervention for abscess drainage or granuloma-related obstruction
- Nutritional support and management of growth failure
- Psychosocial support for patients and families
Gene Therapy and Experimental Treatments
Gene therapy represents a promising frontier in CGD management. Recent advances have enabled the use of viral vectors to deliver functional copies of defective genes into patient-derived haematopoietic stem cells. Early clinical trials have demonstrated the feasibility and potential efficacy of this approach, though long-term safety and durability remain under investigation.
Prognosis
Life Expectancy and Quality of Life
The prognosis for CGD patients has improved significantly over recent decades, largely due to advances in antimicrobial prophylaxis, early diagnosis, and supportive care. Many patients now survive into adulthood, particularly in high-resource settings. Nonetheless, life expectancy is still reduced compared to the general population, and quality of life may be compromised by recurrent infections, chronic inflammation, and treatment-related complications.
Factors Affecting Outcomes
- Patient adherence to prophylactic regimens and follow-up.
- Type of genetic mutation: X-linked forms tend to be more severe than autosomal recessive forms.
- Access to comprehensive medical care and early intervention.
- Success of stem cell transplantation or other curative therapies.
Nursing Care of Patients with Chronic Granulomatous Disease (CGD)
Because of the complex nature of CGD, nursing care requires a holistic and proactive approach, involving infection prevention, early recognition of complications, patient and family education, psychosocial support, and coordination with a multidisciplinary team.
Nursing Assessment
An accurate and thorough assessment is the foundation of effective nursing care for CGD. Key points include:
- Medical and Family History: Document frequency, severity, and types of infections. Inquire about family history of immunodeficiency.
- Current Symptoms: Assess for fever, malaise, cough, skin lesions, gastrointestinal symptoms, or pain indicating possible infection or granuloma formation.
- Physical Examination: Perform a head-to-toe examination, paying special attention to skin, lymph nodes, respiratory system, and abdominal tenderness.
- Laboratory Investigations: Monitor white blood cell counts, inflammatory markers, cultures, and imaging studies as indicated to evaluate for infection or organ involvement.
Infection Prevention and Control
Preventing infection is a nursing priority, as patients with CGD are particularly vulnerable.
- Hand Hygiene: Emphasize and model strict hand hygiene for patients, family, and visitors.
- Isolation Precautions: Implement protective isolation protocols when necessary, especially during hospitalizations or when neutropenic.
- Environmental Controls: Reduce exposure to potential sources of infection, such as fresh flowers, standing water, soil, and crowding.
- Vaccinations: Advocate for appropriate immunizations (avoid live vaccines unless directed by a specialist).
- Antimicrobial Prophylaxis: Ensure adherence to prescribed prophylactic antibiotics (such as trimethoprim-sulfamethoxazole) and antifungals (such as itraconazole).
Early Recognition and Management of Infections
Prompt intervention is crucial in controlling infections and reducing morbidity.
- Monitor for Signs of Infection: Watch for fever, localized pain, swelling, redness, or drainage from wounds.
- Education: Teach patients and families to recognize early signs of infection and seek medical attention promptly.
- Timely Treatment: Ensure that any suspected infection is evaluated rapidly and treated with appropriate antibiotics or antifungals as prescribed.
- Wound Care: Maintain strict aseptic technique for dressing changes and wound care.
- Reporting: Communicate changes in condition to the healthcare team without delay.
Management of Granulomas and Complications
Chronic inflammation can result in granuloma formation, potentially causing obstruction or organ dysfunction.
- Assessment: Monitor for signs of gastrointestinal obstruction (vomiting, abdominal pain, constipation) or urinary tract problems (dysuria, retention).
- Supportive Care: Administer prescribed corticosteroids or other immunomodulatory agents as ordered to reduce inflammation and granuloma size.
- Collaborative Management: Work closely with gastroenterology, urology, and other specialists for surgical or procedural interventions if needed.
- Nutritional Support: Assess for malnutrition; collaborate with dietitians to optimize nutrition, especially if gastrointestinal absorption is compromised.
Patient and Family Education
Empowering patients and families is critical for ongoing disease management.
- Understanding CGD: Provide clear, age-appropriate explanations of the disease process, treatment rationale, and prognosis.
- Home Infection Control: Teach hygiene practices, safe food handling, and avoidance of high-risk exposures (e.g., gardening, contact with animals, swimming in lakes).
- Medication Adherence: Educate on the importance of taking prophylactic medications as prescribed, including how to manage side effects.
- When to Seek Help: Give clear instructions on recognizing infection signs and when to contact healthcare providers.
- Resource Referral: Direct families to support groups, genetic counseling, and educational resources.
Psycho-social and Emotional Support
Living with CGD can cause significant emotional and social challenges.
- Counseling: Offer access to counseling services for anxiety, depression, or coping difficulties.
- Peer Support: Encourage participation in support groups for children and families facing similar challenges.
- School and Activity Planning: Collaborate with teachers and coaches to facilitate a safe and inclusive environment, with plans for infection prevention.
- Addressing Stigma: Support open communication to address misconceptions and reduce feelings of isolation or difference.
Coordination of Multidisciplinary Care
Patients with CGD require regular follow-up with multiple healthcare providers.
- Team Communication: Facilitate regular communication among primary care, immunology, infectious disease, pharmacy, and other specialists.
- Monitoring and Follow-up: Track scheduled appointments, laboratory monitoring, and surveillance imaging.
- Preparation for Procedures: Provide pre- and post-procedure care, especially for biopsies, surgical interventions, or infusion therapies.
Advanced Therapies and Transplant Considerations
For some patients, advanced therapies may be indicated.
- Hematopoietic Stem Cell Transplantation (HSCT): Educate families about the potential for cure with HSCT, including risks, benefits, and long-term follow-up needs.
- Gene Therapy: Provide information on clinical trials and advances in gene therapy, which may offer future treatment options.
- Preparation and Support: Prepare patients and families for intensive treatments, hospitalizations, and possible complications.
Discharge Planning and Community Support
A smooth transition from hospital to home is essential for optimal outcomes.
- Discharge Education: Reinforce infection prevention, medication management, and emergency contacts.
- Home Health Services: Arrange for home nursing support if needed for wound care, medication administration, or monitoring.
- Community Resources: Link patients to community resources, such as social workers, support organizations, and transportation services.
- School Reintegration: Collaborate with educators to support a safe and supportive return to school.
REFERENCES
- Zerbe CS, et al. Chronic granulomatous disease: Pathogenesis, clinical manifestations, and diagnosis. https://www.uptodate.com/contents/search.
- Genetic and Rare Diseases Information Center (U.S.). Chronic granulomatous disease. https://rarediseases.info.nih.gov/diseases/6100/chronic-granulomatous-disease. Last reviewed 2/2023.
- Keller MD, et al. Future of care for patients with chronic granulomatous disease: Gene therapy and targeted molecular medicine. Journal of the Pediatric Infectious Diseases Society. 2018; doi:10.1093/jpids/piy011.
- National Institute of Allergy & Infectious Diseases. (U.S.). Chronic Granulomatous Disease (CGD) (https://www.niaid.nih.gov/diseases-conditions/chronic-granulomatous-disease-cgd). Last reviewed 5/22/2020.
- Chronic granulomatous disease (CGD). Merck Manual Professional Version. https://www.merckmanuals.com/professional/immunology-allergic-disorders/immunodeficiency-disorders/chronic-granulomatous-disease-cgd
- National Organization for Rare Disorders (U.S.). Chronic Granulomatous Disease. https://rarediseases.org/rare-diseases/chronic-granulomatous-disease/. Last reviewed 8/9/2023.
- Chronic granulomatous disease. Medline Plus. https://ghr.nlm.nih.gov/condition/chronic-granulomatous-disease?_ga=1.168947753.905232672.1468720729.
Stories are the threads that bind us; through them, we understand each other, grow, and heal.
JOHN NOORD
Connect with “Nurses Lab Editorial Team”
I hope you found this information helpful. Do you have any questions or comments? Kindly write in comments section. Subscribe the Blog with your email so you can stay updated on upcoming events and the latest articles.


