


Last Updated on January 11, 2026 by Nurseslab.in Editorial Team
Cardiac amyloidosis is a rare but increasingly recognized cause of heart disease, characterized by the deposition of abnormal protein fibrils called amyloid in the heart tissue. This disorder can lead to significant cardiac dysfunction, affecting quality of life and overall survival. Understanding cardiac amyloidosis requires an exploration of its mechanisms, types, clinical manifestations, diagnosis, and management strategies.
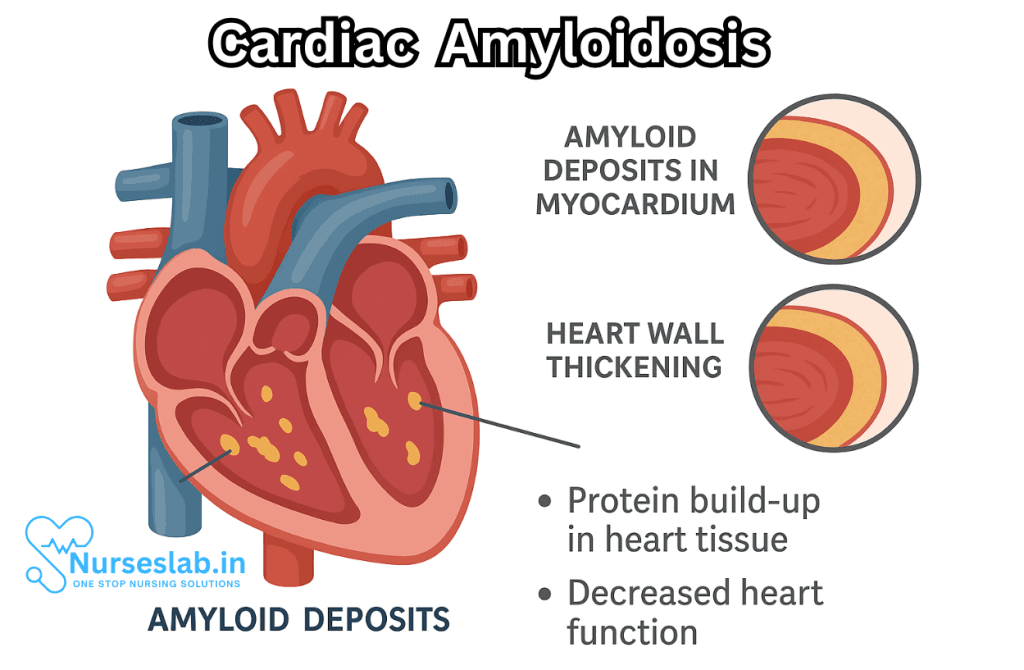
What is Cardiac Amyloidosis?
Cardiac amyloidosis is part of a broader group of diseases known as amyloidoses, in which misfolded proteins accumulate as insoluble fibrils in tissues and organs. When this process involves the heart, it leads to stiffening of the cardiac walls, restrictive cardiomyopathy, and progressive heart failure. The heart’s ability to fill and pump blood efficiently is steadily compromised.
Types of Cardiac Amyloidosis
There are several forms of amyloidosis, but two main types account for most cases of cardiac involvement:
- AL (Light Chain) Amyloidosis: Caused by abnormal plasma cells in the bone marrow producing excess immunoglobulin light chains, which misfold and deposit as amyloid fibrils in tissues.
- ATTR (Transthyretin) Amyloidosis: Linked to the misfolding of transthyretin, a liver-produced protein that normally transports thyroxine and retinol. ATTR amyloidosis is further divided into:
- Hereditary (mutant) ATTR amyloidosis: Due to mutations in the transthyretin gene.
- Wild-type (senile) ATTR amyloidosis: Occurs without mutations, usually in older adults.
Other forms, such as AA (secondary) amyloidosis, rarely involve the heart.
Causes and Risk Factors
Amyloidosis is fundamentally a disorder of protein folding. The causes depend on the specific type:
- AL amyloidosis is associated with plasma cell dyscrasias, such as multiple myeloma or monoclonal gammopathy of undetermined significance (MGUS).
- Hereditary ATTR amyloidosis follows an autosomal dominant inheritance pattern and can manifest at varying ages, depending on the mutation.
- Wild-type ATTR amyloidosis typically affects elderly individuals, particularly men over 65 years old.
Risk factors include age, family history, chronic inflammatory conditions (for AA amyloidosis), and pre-existing plasma cell disorders.
Pathophysiology
The process begins with the misfolding of specific proteins, which aggregate into amyloid fibrils. These fibrils deposit in the myocardial interstitium, replacing healthy tissue and resulting in:
- Increased ventricular wall thickness without true hypertrophy
- Ventricular stiffening and impaired diastolic filling
- Progressive decline in systolic function
- Disruption of the cardiac conduction system, leading to arrhythmias and heart block
Ultimately, these changes culminate in restrictive cardiomyopathy and heart failure.
Clinical Manifestations
Cardiac amyloidosis often presents with non-specific symptoms, making early diagnosis challenging. Common features include:
- Progressive shortness of breath, especially with exertion
- Fatigue and reduced exercise tolerance
- Swelling of the legs and ankles (peripheral edema)
- Orthostatic hypotension (dizziness upon standing)
- Palpitations or syncope (fainting), due to arrhythmias
- Chest discomfort or angina (less common)
Extra-cardiac manifestations may include carpal tunnel syndrome, peripheral neuropathy, macroglossia (enlarged tongue), periorbital purpura, and unexplained weight loss, particularly in AL amyloidosis.
Diagnosis
Diagnosing cardiac amyloidosis requires a high degree of clinical suspicion and a combination of tests:
- Electrocardiogram (ECG): May reveal low voltage in limb leads or conduction disturbances.
- Echocardiography: Shows increased ventricular wall thickness with a characteristic ‘sparkling’ or granular myocardial appearance and diastolic dysfunction.
- Cardiac Magnetic Resonance (CMR): Highly sensitive for detecting amyloid infiltration, with late gadolinium enhancement patterns.
- Laboratory tests: Serum and urine protein electrophoresis, immunofixation, serum free light chains (for AL amyloidosis); genetic testing for suspected hereditary ATTR.
- Nuclear imaging: Bone scintigraphy using technetium-labeled tracers can differentiate ATTR from AL amyloidosis.
- Tissue biopsy: The gold standard. Congo red staining under polarized light reveals apple-green birefringence. Biopsy may be obtained from the endomyocardium or less invasive sites like abdominal fat pad or minor salivary glands.
An accurate diagnosis is crucial as it guides therapy and prognosis.
Treatment Approaches
Treatment is determined by the type of amyloidosis and focuses on two main goals: halting further amyloid deposition and managing heart failure symptoms.
Treating AL Amyloidosis
The cornerstone is suppression of the abnormal plasma cell clone:
- Chemotherapy regimens similar to those used in multiple myeloma (e.g., bortezomib, cyclophosphamide, dexamethasone)
- Autologous stem cell transplant in selected patients
- Supportive cardiac care
Novel therapies, such as monoclonal antibodies targeting amyloid fibrils (e.g., daratumumab), are under investigation.
Treating ATTR Amyloidosis
Therapies aim to stabilize or reduce transthyretin amyloid formation:
- Tafamidis: A transthyretin stabilizer, approved for ATTR cardiomyopathy, improves survival and quality of life.
- Patisiran and inotersen: RNA interference and antisense therapies that reduce transthyretin production, effective primarily for hereditary ATTR with neuropathy but may benefit cardiac disease.
- Liver transplantation: For hereditary cases, as the liver produces mutant transthyretin.
- Supportive management including diuretics, cautious use of beta-blockers, and antiarrhythmic agents
Heart transplantation is considered for refractory cases in eligible patients.
Managing Heart Failure and Arrhythmias
Heart failure in cardiac amyloidosis is particularly challenging:
- Salt restriction and careful use of diuretics to manage fluid overload
- Mineralocorticoid receptor antagonists may be used
- ACE inhibitors, ARBs, and beta-blockers are often poorly tolerated
- Implantable devices (pacemakers, defibrillators) for significant conduction disease or ventricular arrhythmias
Regular monitoring and a multidisciplinary approach are essential.
Nursing Care of the Patient with Cardiac Amyloidosis
Due to the chronic and progressive nature of cardiac amyloidosis, nursing care plays a crucial role in improving quality of life, minimising symptoms, and supporting patients and families throughout their healthcare journey.
Understanding Cardiac Amyloidosis
To deliver optimal nursing care, it is essential to understand the two primary types of cardiac amyloidosis:
- AL (Light Chain) Amyloidosis: Arises from abnormal plasma cells producing misfolded immunoglobulin light chains, which deposit as amyloid fibrils. Cardiac involvement can be aggressive and often presents a poorer prognosis if untreated.
- ATTR (Transthyretin) Amyloidosis: Results from misfolded transthyretin proteins, including hereditary (mutant) and wild-type forms. ATTR amyloidosis generally progresses more slowly, and new therapies are improving outcomes for affected patients.
Assessment and Monitoring
Thorough assessment and vigilant monitoring are foundational to nursing care for cardiac amyloidosis. Nurses must recognise subtle changes in clinical status, as these can rapidly progress and warrant urgent intervention.
Baseline Assessment
- History and Symptoms: Document cardiac symptoms such as dyspnoea, orthopnoea, palpitations, syncope, and signs of congestive heart failure. Assess for fatigue, peripheral oedema, and gastrointestinal complaints.
- Physical Examination: Monitor vital signs, jugular venous pressure, breath sounds, heart sounds (noting murmurs or arrhythmias), peripheral pulses, and presence of oedema.
- Functional Status: Evaluate exercise tolerance, ability to perform activities of daily living, and cognitive function. Use validated tools such as the NYHA classification or 6-minute walk test where appropriate.
Ongoing Monitoring
- Regularly assess weight, fluid balance, and urine output to detect worsening heart failure.
- Monitor laboratory markers: B-type natriuretic peptide (BNP), troponins, renal function, and electrolytes.
- Observe for arrhythmias or conduction disturbances using ECG monitoring, especially in patients with pacemakers or defibrillators.
- Track response to therapies and report any new or worsening symptoms promptly.
Management of Cardiac Symptoms
Patients with cardiac amyloidosis are prone to heart failure, arrhythmias, and conduction abnormalities. Nursing interventions focus on symptom relief, preventing complications, and supporting medical management.
Heart Failure Management
- Administer prescribed medications carefully, noting that ACE inhibitors, ARBs, and beta-blockers may be poorly tolerated. Monitor for hypotension, renal dysfunction, and electrolyte imbalances.
- Support the use of mineralocorticoid receptor antagonists and diuretics, observing for dehydration and orthostatic hypotension.
- Monitor fluid intake and output. Educate patients on fluid and sodium restriction if advised by the clinical team.
- Assess for signs of pulmonary oedema and promptly escalate care if required.
Arrhythmia and Conduction Disease Management
- Monitor heart rate and rhythm closely; administer antiarrhythmic therapy as prescribed.
- Provide care and education for patients with pacemakers or defibrillators, including device site assessment and troubleshooting alarms.
- Prepare patients and families for possible interventions such as device implantation or electrophysiological studies.
Patient Education and Self-Management
Empowering patients with knowledge about their condition fosters better self-management and emotional resilience.
- Educate patients and families about cardiac amyloidosis, its symptoms, treatment options, and prognosis.
- Teach recognition of warning signs such as increased shortness of breath, rapid weight gain, swelling, chest pain, or palpitations. Encourage prompt reporting of these symptoms.
- Explain medication regimens, including potential side effects and the importance of adherence. Address misconceptions and answer questions to reduce anxiety.
- Guide lifestyle modifications: low-sodium diet, gentle physical activity as tolerated, smoking cessation, and avoidance of alcohol or recreational drugs.
Psychosocial and Emotional Support
A diagnosis of cardiac amyloidosis can be overwhelming. Nurses serve as advocates, emotional supports, and resources for patients and families.
- Assess psychological well-being and screen for depression, anxiety, and coping challenges. Provide referrals to counselling or psychiatric services as needed.
- Encourage participation in patient advocacy groups and support networks, which offer education, peer support, and connection to current research advances.
- Facilitate communication between patients, families, and the multidisciplinary team, helping to clarify goals of care and treatment expectations.
- Respect cultural beliefs, values, and preferences in decision-making and care planning.
Multidisciplinary Collaboration
Optimal outcomes for cardiac amyloidosis are achieved through a multidisciplinary approach.
- Coordinate care with cardiologists, haematologists, palliative care specialists, physiotherapists, dietitians, pharmacists, and social workers.
- Participate in regular case conferences and care planning meetings.
- Ensure continuity of care between inpatient and outpatient settings, arranging follow-up appointments and facilitating transitions.
Recent Advances and Research
Nurses should stay abreast of emerging therapies and clinical trials, which may offer new hope for patients.
- Gene-silencing therapies (e.g., patisiran, inotersen) and novel agents targeting amyloid fibrils are transforming ATTR amyloidosis management.
- Advanced imaging techniques, such as cardiac MRI and nuclear scans, improve early detection and disease monitoring.
- Ongoing clinical trials are evaluating additional therapeutic options, and nurses can educate and encourage eligible patients to consider participation.
Living with Cardiac Amyloidosis: The Nursing Perspective
- Nurses are integral in supporting patients in their day-to-day lives with cardiac amyloidosis.
- Encourage regular follow-up and adherence to care plans.
- Help patients adapt their routines to changing functional status and disease progression.
- Address symptoms proactively, promoting comfort and dignity.
REFERENCES
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2021;42(16):1554-1568. doi:10.1093/eurheartj/ehab072
- Bustamante JG, Zaidi SRH. Amyloidosis. https://www.ncbi.nlm.nih.gov/books/NBK470285/). [Updated 2021 Aug 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-.
- Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4820353/. Cell Mol Life Sci. 2009;66(19):3095-3101.
- Estep JD, Bhimaraj A, Cordero-Reyes AM, Bruckner B, Loebe M, Torre-Amione G. Heart transplantation and end-stage cardiac amyloidosis: a review and approach to evaluation and management. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3487570/. Methodist Debakey Cardiovasc J. 2012;8(3):8-16.
- Teng C, Li P, Bae JY, Pan S, Dixon RAF, Liu Q. Diagnosis and treatment of transthyretin-related amyloidosis cardiomyopathy. Clin Cardiol. 2020;43(11):1223-1231. doi:10.1002/clc.23434
- Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. https://pubmed.ncbi.nlm.nih.gov/29700090/. Clin Med (Lond). 2018;18(Suppl 2):s30-s35.
- National Organization for Rare Diseases | rarediseases.org. Amyloidosi.. https://rarediseases.org/rare-diseases/amyloidosis/.
- Sachchithanantham S, Wechalekar AD. Imaging in systemic amyloidosis. https://pubmed.ncbi.nlm.nih.gov/23896486/. Br Med Bull. 2013;107:41-56.
- Baker KR. Light chain amyloidosis: Epidemiology, staging, and prognostication. Methodist Debakey Cardiovasc J. 2022;18(2):27-35. doi:10.14797/mdcvj.1070
- Scarpioni R, Ricardi M, Albertazzi V, et al. Dialysis-related amyloidosis: challenges and solutions. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5153266/. Int J Nephrol Renovasc Dis. 2016;9:319-328. Published 2016 Dec 7.
- U.S. National Library of Medicine | MedlinePlus. Multiple pages reviewed relating to cardiac amyloidosis. https://medlineplus.gov/ency/article/000193.htm.
Stories are the threads that bind us; through them, we understand each other, grow, and heal.
JOHN NOORD
Connect with “Nurses Lab Editorial Team”
I hope you found this information helpful. Do you have any questions or comments? Kindly write in comments section. Subscribe the Blog with your email so you can stay updated on upcoming events and the latest articles.


